you may also like

In the rapidly maturing landscape of 2026, T cell engager (TCE) therapies have transitioned from a promising modality to a cornerstone of modern immunotherapy. With a global market value reaching approximately $7.7 billion in 2025 and a projected growth rate exceeding 15% annually, the race to expand target landscapes across hematologic and solid tumors is accelerating. However, as these therapies grow in complexity, the industry faces a critical bottleneck: the need for pharmacological resolution that current in vivo models struggle to provide.
T cell engagers (TCEs) act as molecular “middlemen”, requiring precise bridging of human T cells to targeted cells. This makes their preclinical evaluation highly dependent on how accurately animal models reproduce these human immune interactions.
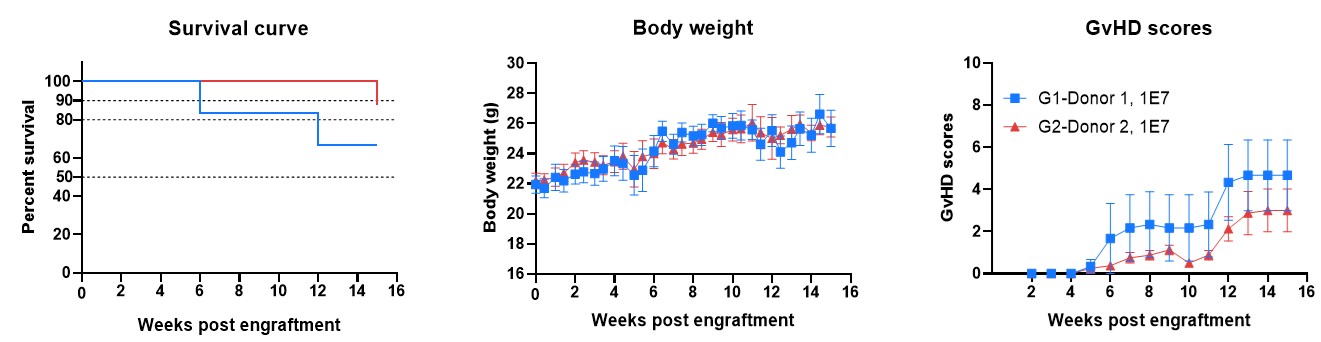
A major limitation lies in the major histocompatibility complex (MHC) system—specifically MHC class I and II—which regulates antigen presentation and T cell recognition of “self” versus “non-self”. In human PBMC-engrafted mouse models, murine MHC molecules can aberrantly activate human T cells, introducing a fundamental cross-species incompatibility—the “immune noise”. Although these models are widely used due to rapid and convenient human T cell reconstitution, this incompatibility also drives human T-cell-mediated graft-versus-host disease (GvHD), leading to premature death of the animals and severely constraining study duration and interpretability.
This incompatibility results in a distorted immune environment characterized by:
Together, these factors significantly reduce the predictive power of conventional PBMC-humanized models, particularly for next-generation TCEs where precise immune activation is critical.
The tarlatamab analog (in-house) demonstrates potent, dose-dependent tumor inhibition in the SHP-77 model while maintaining a superior safety profile; unlike the anti-hCD3 control (G5)—which triggers indiscriminate, off-target T-cell activation throughout the body—the analog restricts immune activity to the tumor site, preserving animal health (Figure 2).
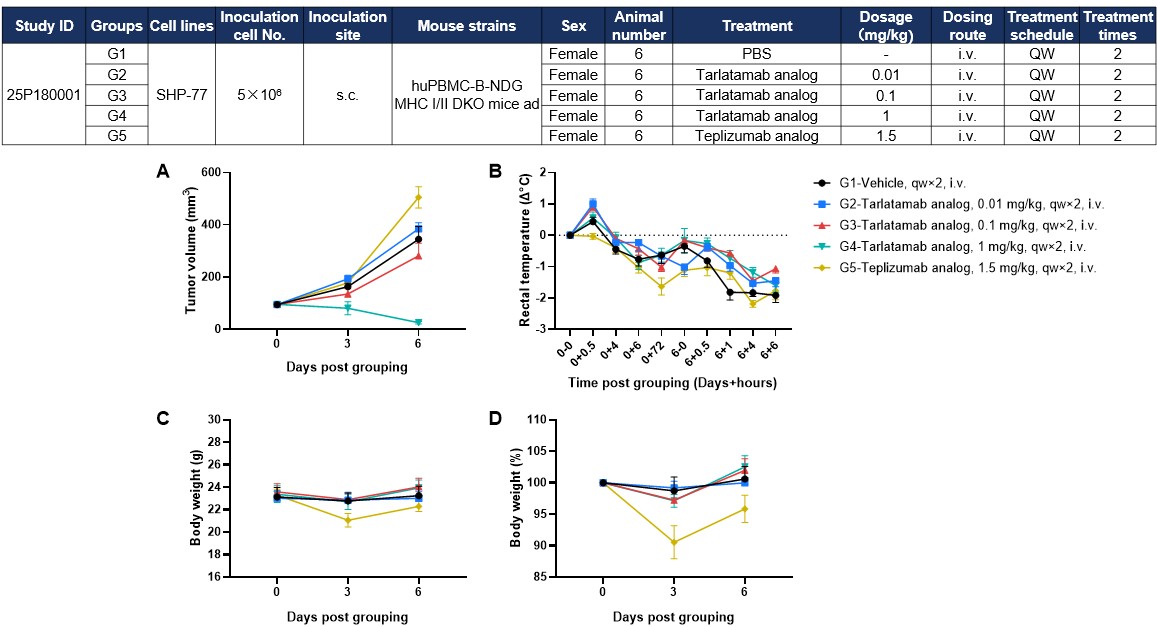
Figure 2. Antitumor Activity and Toxicity of Anti-Human CD3/DLL3 Bispecific T-Cell Engager (Tarlatamab Analog, In-House) in Small Cell Lung Cancer Cell Line SHP-77 CDX Model Established With hPBMC-B-NDG MHC I/II DKO Mice Ad. Human PBMCs (1×107) were intravenously engrafted into B-NDG MHC I/II DKO mice ad, followed by a study design on the top. (A) Tarlatamab analog could effectively inhibit tumor growth in a dose-dependent manner, whereas teplizumab analog showed no tumor antigen–dependent antitumor activity. (B–D) Teplizumab analog induced pronounced body weight loss and hypothermia, while tarlatamab analog caused only mild body weight changes with less severe decreases in rectal temperature. Data are presented as mean ± SEM.
By eliminating non-specific immune noises, the model ensures that plasma cytokine levels (such as IL-6, IFN-γ, and TNF-α) are an accurate reflection of the therapeutic's activity rather than host-driven inflammation. This resolution allowed researchers to identify that cytokine release was more pronounced following the first administration than after the second dose, providing critical pharmacological insights for clinical trial dosing strategies (Figure 3).
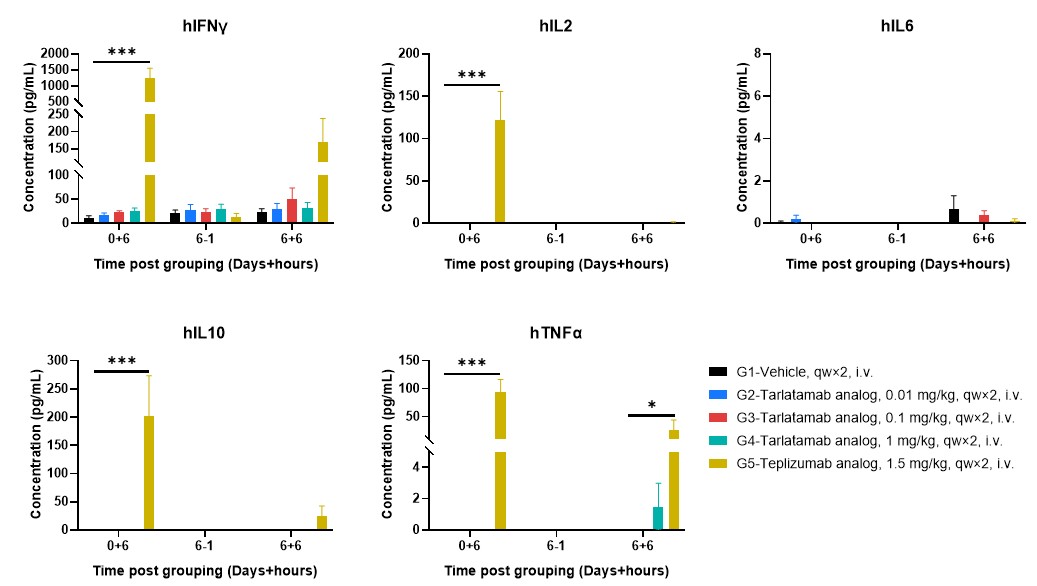
Figure 3. Plasma Cytokine Releases Profiling after First and Repeat Dosing in hPBMC-B-NDG MHC I/II DKO Mice Ad. Plasma cytokine levels were assessed at defined time points following dosing, including 6 hours after the first administration (0+6), one hour prior to the second administration on day 6 (6−1), and 6 hours after the second administration (6+6). Teplizumab analog-treated mice exhibited elevated cytokine levels, whereas cytokine levels in tarlatamab analog-treated groups were low or below the detection range at the indicated time points. Data are presented as mean ± SEM. Significance was determined by two-way ANOVA test. *P < 0.05, **P < 0.01, ***P < 0.001.
The B-NDG MHC I/II DKO Mice Ad supports the sustained engraftment and persistence of human CD45+ and CD3+ cells with stable CD4/CD8 ratios across peripheral blood (Figure 4), spleen, and tumor tissues (Figure 5). This stability ensures that increases in T-cell absolute counts and the upregulation of activation markers like CD69 are driven by specific engagement with the therapeutics and the tumor antigen, rather than artificial activation by murine MHC molecules.
Tarlatamab treatment maintains high human CD3+ T-cell frequencies and stable CD4/CD8 compositions in the peripheral blood (Figure 4), spleen (Figure 5A), and tumor (Figure 5B). Higher doses (1 mg/kg) promote a clear increase in circulating human leukocyte and T-cell counts while driving target-specific, rather than host-driven, immune activation demonstrated by elevated CD69.
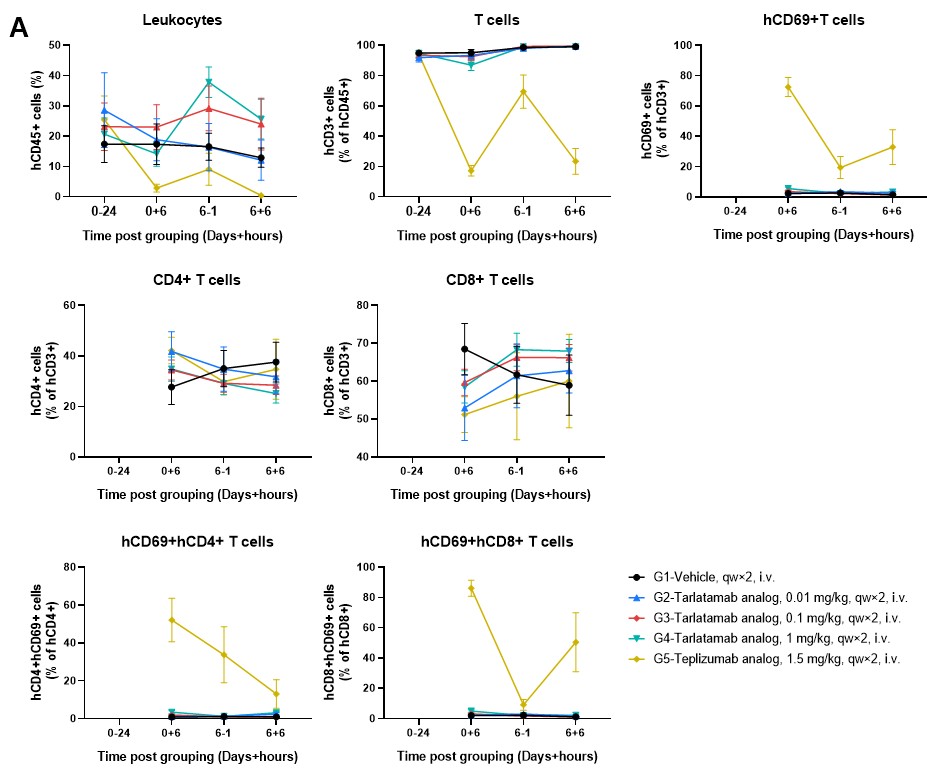
Figure 4. Longitudinal Analysis of Peripheral Human Immune Cell Dynamics in hPBMC-B-NDG MHC I/II DKO Mice Ad. (A) Frequency of human leukocytes (hCD45+), T cells (hCD3+), and activated T cells (hCD69+) expressed as a percentage of their respective parent populations. (B) Absolute counts (cells/μL) of the same immune subsets. Teplizumab causes acute, systemic T cell activation followed by depletion, whereas the Tarlatamab analog (particularly at 1 mg/kg) promotes a progressive expansion of the T cell pool with more controlled activation kinetics, likely reflecting T cell proliferation and recruitment rather than a systemic activation “burst”. Data are presented as mean ± SEM.
Figure 5. The Frequencies of Human Immune Cell Populations in Spleen and Tumor Tissue of hPBMC-B-NDG MHC I/II DKO Mice Ad. Immune cells from (A) spleens and (B) tumor tissues were analyzed using flow cytometry. Data are presented as mean ± SEM. Significance was determined by one-way ANOVA test. *P < 0.05, **P < 0.01, ***P < 0.001.
As TCE therapies continue to evolve and expand into solid tumors and autoimmune indications, the distinction between "good enough" data and "clinically actionable" data becomes vital. By improving immune stability and extending the experimental window, our B-NDG MHC I/II DKO Mice Ad provides a reliable, translationally relevant platform that helps researchers move past the noise and toward the next breakthrough.
Is your current model providing a clear signal—or adding more noise? Explore how Biocytogen’s advanced immune reconstitution models can refine your TCE pipeline. Contact us to learn more!
Our model eliminates murine MHC class I and II, preventing mouse antigen presentation. This reduces xenogeneic T-cell activation and allows drug-driven immune responses to be evaluated more cleanly.
TCEs activate T cells via CD3. In standard models, murine MHC can non-specifically activate human T cells, creating background noise. Removing MHC ensures that T-cell activation is primarily driven by the therapeutic.
Yes. Our model is particularly valuable for: